Sprache ändern Englisch
Übersicht gemäss der revidierten IC3D Klassifikation 2015
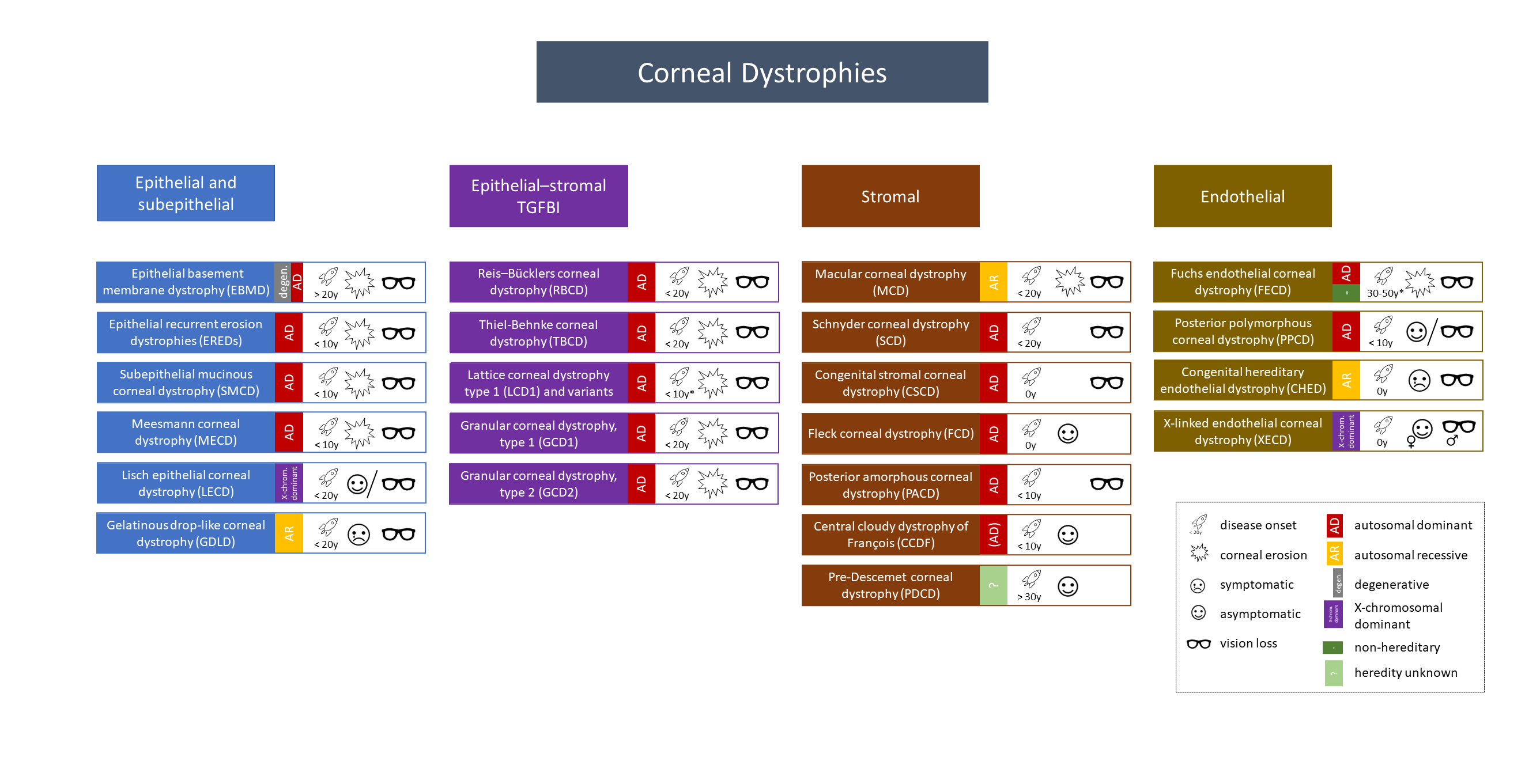
Allgemeines
- erbliche, bilaterale, progressive Hornhaut-Erkrankungen
- Ausnahmen vorhanden
- Familienanamnese erheben
- Untersuch: immer in Mydriase, immer Partnerauge untersuchen
- Kategorien
- C1: klinisch und histologisch klar definierte Dystrophie mit Identifikation des Gens und der Mutationen
- C2: klinisch und histologisch klar definierte Dystrophie mit bekannter Chromosomenlokalisation und unbekannter Genidentifikation
- C3: klinisch und histologisch klar definierte Dystrophie ohne genetische Analyse
- C4: Verdacht auf eine neue oder bereits dokumentierte Dystrophie, wobei die Eigenständigkeit noch nicht erwiesen ist
- Merkspruch: Marilyn Monroe Always Gets Her Men in L. A. County
- Macular dystrophy – Mucopolysaccharide – Alcian blue
- Granular dystrophy – Hyaline materials – Masson trichrome
- Lattice dystrophy – Amyloid – Congo red
Epitheliale und subepitheliale Dystrophien
Epitheliale Basalmembrandystrophie (EBMD)
- siehe separater Artikel: Map-Dot-Fingerprint-Dystrophie
Epitheliale rezidivierende Erosionsdystrophien (EREDs)
- Allgemeines: Beginn in 1. Lebensjahrzehnt (meist 4-6-jährig), autosomal dominant, C4
- Variante: Dystrophia Smolandiensis C3
- Klinik
- Rezidivierende Erosionen in Kindheit meist ohne Trübungen
- Anfälle mit Rötung, Photophobie, Epiphora und Augenschmerzen, Augenbrennen, empfindliche Augen über mehrere Jahre
- induzierbar durch Sonnenlicht, Zugluft, Staub, Rauch und Schlafmangel
- erwachsene Patienten mit Smolandiensis Variante haben zu 50% zentrale diffuse, subepitheliale Trübungen, zT Keloid-artig
- 25% mit Hornhauttransplantat Mitte 40 wegen Visusminderung
- meist mit 50 Jahren keine Schmerz-Attacken mehr
Subepitheliale muzinöse Hornhautdystrophie (SMCD)
- Allgemeines: Beginn in 1. Lebensjahrzehnt, autosomal dominant, C4 (gehört wahrscheinlich zu EREDs)
- Klinik
- Rezidivierende Erosionen in Kindheit
- bei Erwachsenen diffuse subepitheliale Trübungen und Haze
- Progredienter Visusverlust in Pubertät
Meesmann-Hornhautdystrophie (MECD)
- Allgemeines: Beginn in früher Kindheit, autosomal dominant, C1
- Klinik
- häufig asymptomatisch, leichte Visusminderungen, gelegentlich punktförmige Erosionen
- diffuse, graue Trübungen in direkter Beleuchtung
- multiple, solitäre, runde und klare Epithelbläschen in indirekter Beleuchtung, zT bis zum Limbus
- langsam progredient
Lisch epitheliale Hornhautdystrophie (LECD)
- Allgemeines: Beginn in Kindheit, X-chromosomal-dominant, C2,
- Klinik
- kein Unterschied zwischen Männer und Frauen!
- asymptomatisch oder Visusminderung bei zentralen Veränderungen, keine Erosionen!
- diffuse graue Trübungen in direkter Beleuchtung
- multiple, zusammengeballte, runde und klare Mikrozysten in indirekter Beleuchtung
- langsame Progredienz
Gelatinöse tropfenförmige Hornhautdystrophie (GDLD)
- Allgemeines: Beginn in 1.-2. Lebensjahrzehnt, autosomal rezessiv, C1
- Klinik:
- starke Visusminderung, Photophobie, Reizung, Rötung, Tränen
- subepithelialer Veränderungen (Bandkeratopathietyp), am häufigsten
- multiple kleine maulbeerförmige Knötchen (Maulbeer-Typ), verzögerte Anfärbung mit Fluorescein (durchlässiges Epithel), häufig oberflächliche Vaskularisation
- in späteren Jahren Stromatrübung mit grösseren nodulären Läsionen („Kumquat-Typ“)
- Progredienz, Rezidiv innert weniger Jahren nach oberflächlicher Keratektomie oder Hornhauttransplantation
Epithelial-stromale TGFBI Dystrophien
Reis-Bücklers-Hornhautdystrophie (RBCD)
- Allgemeines: Beginn in Kindheit, autosomal-dominant, C1, zT schwierig von TBCD zu unterscheiden
- Klinik
- häufig rezidivierende Erosionen (häufiger und heftiger als TBCD), werden schwächer zum Erwachsenenalter hin
- langsam fortschreitende Visusminderung
- subepitheliale, landkartenförmige Trübungen, können sich zum Limbus und Stroma ausdehnen
Thiel-Behnke Hornhautdystrophie (TBCD)
- Allgemeines: Beginn in Kindheit, autosomal-dominant, C1/C2, z.T. schwierig von RBCD zu unterscheiden
- Klinik
- rezidivierende Erosionen, schwächer ausgeprägt als RBCD, können mit der Zeit abnehmen
- Beginn der Visusminderung später als bei RBCD
- subepitheliale, honigwabenförmige Trübungen, können sich zum Limbus und Stroma ausdehnen, Peripherie jedoch meist nicht betroffen
Gittrige (Lattice) Hornhautdystrophie Typ 1 (LCD1) und Varianten
- Allgemeines: Beginn im 1. Lebensjahrzehnt, autosomal dominant, C1
- Klinik
- in den ersten 2 Dekaden häufig rezidivierende Erosionen und Visusminderung
- parazentrale, doppelbrechende Gitterlinien, zentrale progrediente diffuse Trübungen, peripher bleibt ca. 1mm frei von Trübungen
- Visusverschlechterung in 4. Lebensjahrzehnt, führt häufig zu Keratoplastik
- LCD-Varianten (Typ IIIa, I/IIIa, IV und polymorphe Amyloidose) treten später auf als Typ 1
- Typ IIIa: Linien dicker von Limbus zu Limbus
- Typ I/IIIa: dünnere Linien
- polymorphe Amyloidose: keine Linien
- Erosionen bei Typ IIIa und I/IIIa, keine Erosionen bei Typ IV und polymorpher Amyloidose
- Sonderfall: ehemals Gelsolin Typ 2 (LCD2) = Meretoja-Syndrom
- generalisierte Amyloidose mit Hornhautbefall, deshalb falsche Bezeichung, keine Hornhaut-Dystrophie
- autosomal dominant, 3.-4. Lebensjahrzehnt, ähnliche Gitterlinien wie bei Lattice Dystrophie, aber mehr peripher vom Limbus ausgehend
- ausgeprägte Dermatochalasis und Lagophthalmus durch kraniale Neuropathie mit Gesichtslähmung, erhöhtes Risiko für POWG, verminderte Hornhautempfindlichkeit, Sehschärfe bis 6. Lebensjahrzehnt normal, vermehrt trockene Augen, Erosionen im fortgeschrittenen Alter
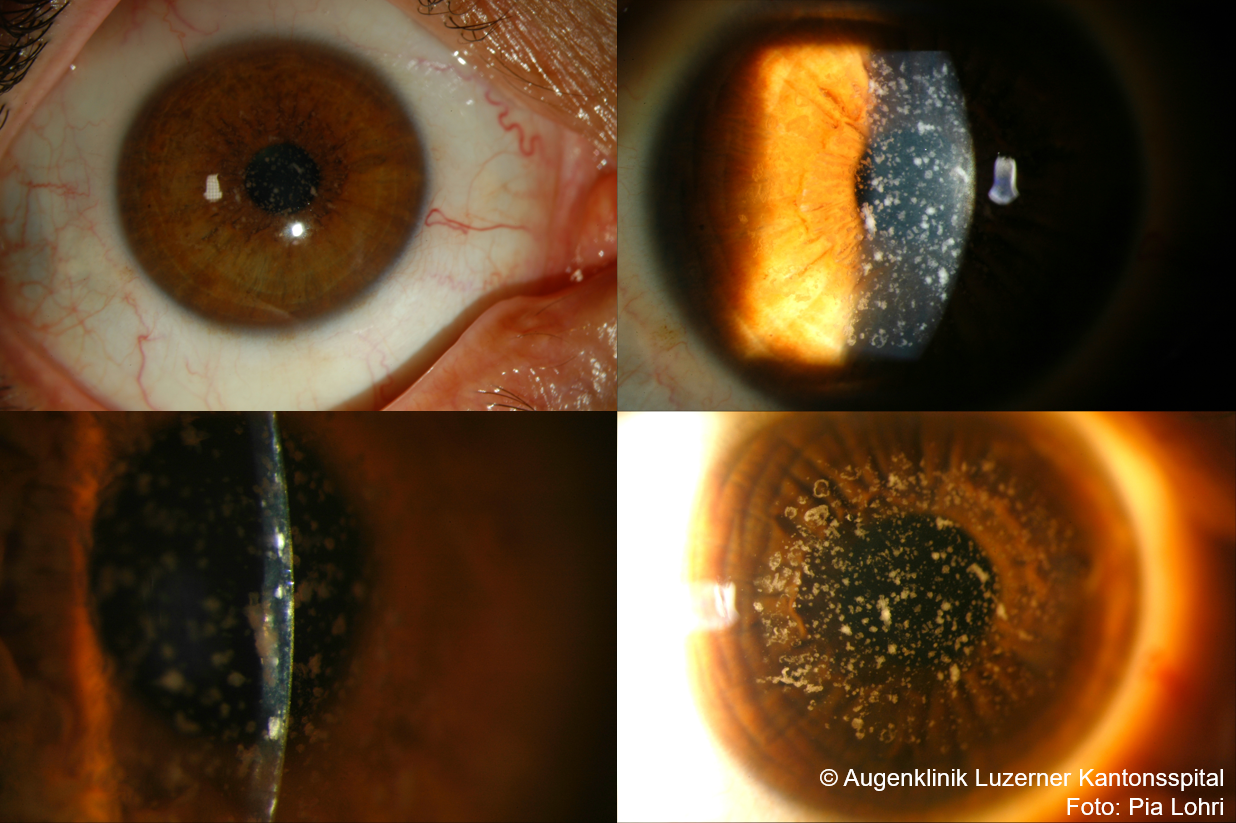
Granuläre Hornhautdystrophie Typ 1 (GCD1)
- Allgemeines: Beginn in Kindheit, autosomal dominant, C1
- Klinik
- Häufig rezidivierende Erosionen, Visusminderung mit progredienten konfluierenden Trübungen im Alter
- zentrale klar abgegrenzte Bröckelchen/Schneegestöber im vorderen Hornhautbereich, weisslich bei direkter Beleuchtung, Trübungen reichen nicht bis Limbus
- klare Hornhaut zwischen Trübungen (im Gegensatz zu makulärer Hornhautdystrophie)
{kind=link}
Granuläre Hornhautdystrophie Typ 2 (GCD2)
- Allgemeines: Beginn in Kindheit, autosomal dominant, C1, wurde früher auch Avellino-Dystrophie genannt
- Klinik
- initial feine weisse zentrale subepitheliale Punkttrübungen, später Ringe und Sterne, häufig in Fingerform, in Spätphase oberflächliche, verbreiterte, brotkrumenartige und teilweise durchscheinende Trübung bis ins vordere/mittlere Stroma
- klare Hornhaut zwischen Trübungen (im Gegensatz zu makulärer Hornhautdystrophie)
- weniger Trübungen als Typ 1
- langsam fortschreitend, im Verlauf Visusminderung durch progrediente zentrale Trübungen, gelegentlich Erosionen
Stromale Dystrophien
Makuläre Hornhautdystrophie (MCD)
- Allgemeines: Beginn in Kindheit, autosomal rezessiv, C1
- Klinik
- starke Visusminderung zwischen 10. und 30. Lebensjahr, rezidivierende Erosionen, Verringerung der Hornhautsensibilität, Photophobie
- zuerst zentrale, später bis in Peripherie und bis Descemet-Membran reichende fleckförmige weissliche Trübungen (Flecken), zwischen Flecken diffuse progrediente Stromatrübung bis zum Limbus (im Gegensatz zu granulärer Hornhautdystrophie)
- häufig Verdünnung der zentralen Hornhaut
- in fortgeschrittenem Stadium Guttae mit Stromaödem möglich
- langsam fortschreitend
Schnyder-Hornhautdystrophie (SCD)
- Allgemeines: Beginn in Kindheit, autosomal dominant, C1, (alter Name: Schnyder’sche kristalline Hornhautdystrophie)
- Diagnosestellung häufig erst in 2.-3. Lebensjahrzehnt (erschwert bei Typ 2 ohne Kristalle)
- Klinik
- Typ 1: zentrale scheiben-/ringförmige Trübung, dicht zusammengeballte, kommaförmige, mehrfarbige subepitheliale Kristalle
- Typ 2: zentrale diffuse, subepitheliale scheiben-/ringförmige Trübung ohne Kristalle
- beide Typen mit Arcus lipoides assoziiert
- im Verlauf Entwicklung einer diffusen Stromatrübung
- photopischer Visus unverhältnismässig stark vermindert bei typischerweise sehr gutem skotopischem Visus
- Langsam progredient, meist ab 50. Lebensjahr Keratoplastik nötig wegen photopischem Visus
Kongenitale stromale Hornhautdystrophie (CSCD)
- Allgemeines: angeboren, autosomal-dominant, C1
- Klinik
- mittelschwere bis schwere Visusminderung
- diffuse, schneeflockenförmige Trübungen in gesamten Hornhautstroma, verdickte Hornhaut
- nicht progredient oder langsam fortschreitend
Fleckchen Hornhautdystrophie (FCD)
- Allgemeines: kongenital, autosomal dominant, C1
- Klinik
- asymptomatisch
- solitäre, schuppenförmige, weisse Stromatrübungen bei ansonsten klarer Hornhaut
- kann asymmetrisch bzw. einseitig sein
- nicht progredient
Posteriore amorphe Hornhautdystrophie (PACD)
- Allgemeines: Beginn in 1. Lebensjahrzehnt, autosomal dominant, C3 (evtl. mesodermale Dysgenesie)
- Klinik
- Visus leicht vermindert, meist > 0.5
- diffuse Trübung mit Aufhellungen v.a. im hinteren Stromabereich
- assoziiert mit Irisanomalien
- nicht progredient oder langsam fortschreitend
Zentral-wolkenförmige Hornhautdystrophie – François (CCDF)
- Allgemeines: Beginn in 1. Lebensjahrzehnt, unbekannt, autosomal dominant (vereinzelt beschrieben), C4
- Klinik
- meist asymptomatisch
- klinisch identisch mit posteriorem Crocodile Chagrin
- zentrale wolkenförmige, krokodillederartige Trübungen im posterioren Stroma, dazwischen klare Hornhaut
- nicht progredient
Prä-Descemet-Hornhautdystrophie (PDCD)
- Allgemeines: Beginn nach 30. Lebensjahr, Vererbung nicht klar, C4
- Klinik
- asymptomatisch, normaler Visus
- punkt-, strich-, ring-, kristalline Trübungen im posterioren Stromabereich
- ähnliche Befunde bei Ichthyosis
- kein klinischer Unterschied zwischen nichtkristalliner PDCD und Cornea farinata
- nicht progredient oder langsam fortschreitend (je nach Form)
Endotheliale Dystrophien
Fuchs-endotheliale Hornhautdystrophie (FECD)
- siehe separater Artikel: Fuchs Endotheldystrophie
Hintere polymorphe Hornhautdystrophie (PPCD)
- Allgemeines: Beginn in früher Kindheit, autosomal-dominant, C1-2
- Klinik
- oft asymptomatisch, bei Fortschreiten in Richtung Stroma deutliche Visusminderung möglich
- Endothel verhält sich wie Epithel
- knötchen-, bläschen-, bandförmige Trübungen, einzeln oder in Gruppen, im posterioren Hornhautbereich
- typische Eisenbahnschienen-ähnliche Veränderungen
- oft jahrelang keine Veränderung der endothelialen Befunde, langsame Progression möglich
Kongenitale hereditäre Endotheldystrophie (CHED)
- Allgemeines: meist kongenital, autosomal-rezessiv, C1
- Bemerkung: wurde früher in CHED1 und CHED2 unterteilt: obsolet, da CHED1=PPCD
- Klinik
- deutliche Visusminderung, Photophobie, geringe Epiphora
- diffuse, milchglasartige Hornhauttrübungen, ggf. kombiniert mit vereinzelten grauen Flecken
- deutlich verdickte Hornhaut (bis 2-3x!)
- häufig Nystagmus
- meist stabiler Verlauf
X-gebundene Endothel-Hornhautdystrophie (XECD)
- Allgemeines: kongenital, X-chromosomal dominant, C2
- bei kongenitaler Hornhautveränderung Untersuchung der Eltern auf endotheliale Veränderung (DD: kongenitales Glaukom!)
- Klinik
- Männer: Visusminderung, milchglasartige Trübung der Hornhaut, Mondkrater-artige endotheliale Veränderungen, sekundäre subepitheliale Bandkeratopathie, evtl. Nystagmus, progredient
- Frauen: asymptomatisch, mondkraterartige endotheliale Veränderungen, meist nicht progredient
Quellen
- Weiss JS, Møller HU, Aldave AJ, Seitz B, Bredrup C, Kivelä T, Munier FL, Rapuano CJ, Nischal KK, Kim EK, Sutphin J, Busin M, Labbé A, Kenyon KR, Kinoshita S, Lisch W. IC3D classification of corneal dystrophies–edition 2. Cornea. 2015 Feb;34(2):117-59. doi: 10.1097/ICO.0000000000000307. Erratum in: Cornea. 2015 Oct;34(10):e32. PMID: 25564336.
- Lisch, Walter & Seitz, Berthold. (2011). Neue internationale Klassifikation der Hornhautdystrophien. Der Ophthalmologe. 108. 883-897. 10.1007/s00347-011-2388-8.
- Seitz B, Lisch W, Weiss J, Die revidierte neueste IC3D-Klassifikation der Hornhautdystrophien; Klin Monatsbl Augenheilkd 2015; 232: 283-294, DOI 10.1055/s-0041-100774
- Cornea-Society – Publications: IC3D Klassifikation in verschiedenen Sprachen zum Downloaden